Topspin contains a very powerful tool for automation – Python. If you aren’t familiar with python, what you should know is that it is extremely easy to pick up, it generalizes very well, and is coding is done ‘in plain english’. By ‘plain english’ I mean that it is very simple to get started compared to other coding languages. In the interests of time, I won’t be going into python, there have been countless tutorials on that posted to youtube, and I simply can’t compete or offer insight into anything better than they can.
Assuming you’ve used Python, you’re in great shape to begin processing your data through topspin. Luckily, topspin installs a native python so that you can begin working with it without having to pull yourself into ‘which python’ territory.
Let’s get started!
Commands are typed in bold - edpy/edmac
Parameters are in italics - TD/SI/LPbin
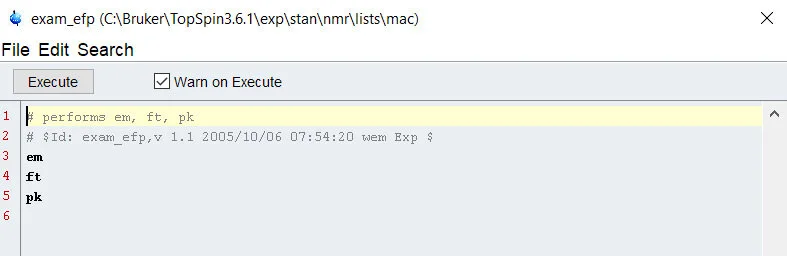
First, we need to open the python module in Topspin. We can do this by simply typing ‘edpy’ into the command line. This brings up a menu allowing you to see some sample python scripts that Bruker has included. For now, lets pull one up and take a look at how we may be able to use this. Perhaps one of the best scripts to get you started would be the ‘py-test-suite’ script.
In this script, we can see that we define some dialog boxes – but importantly, these dialog boxes are defined using topspin specific commands that do not exist in standard python and there are many other commands that are hard coded into topspin python. Bruker’s team has done a lot of work for you already!
As is the case with the last lesson, we are going to design a python script to automate something we already do – in this case we’ll be performing some linear prediction and zero filling, reprocessing the data, and peak picking. For this demonstration, we are going to use linear prediction in both dimensions, although this is usually not done since we can normally acquire enough points in F2 to not need it.
Some basic steps still apply, we are going to create a new python script for this, but in order to do so, you must switch the python module directory to the' “…../py/user” directory in the edpy menu. Once there, select new and give your new masterpiece a title.
This opens up a built in notepad that we can code in and execute the script from directly. This is useful for testing whether a command works or not, so I constantly click that execute button. You’ll notice the “Warn on Execute” button there…I’d disable it when troubleshooting, since it sends a pop-up your way every time you test the code out.
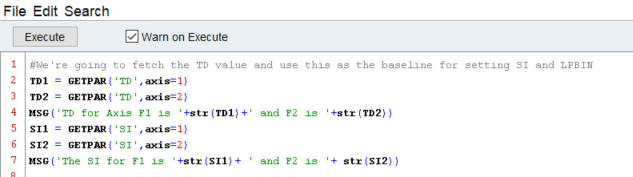
In this first chunk of code, we are simply retrieving the values for the parameter TD from the OPEN dataset. Next, we use a built in dialog box to send a message to the user about what those parameters actually are, and then do the same for SI. You may notice that I tell the script to convert the value of TD1 and TD2 to strings using the str() command. This is actually not required, since the GETPAR( ) function returns values as strings, but I choose to force the hand.
Using these values, we then simply multiply the TD by 2 to find a viable SI value which will allow us to do both linear prediction and zero filling. In order to do this, however, we need to remember that the value of TD1 and TD2 are given as strings - so we tell python to convert that string into an integer. Here, you notice that when we are setting the value, I’ve changed the convention from GETPAR(parameter,axis=XYZ) to PUTPAR(‘axis# variable’, value). You can retrieve the values from GETPAR using this convention as well if you desire.

Once our new SI values are set, we want to tell the system that we plan on using linear prediction, and what kind. We do this by setting the ME_mod parameter to “LPfr'“ (which stands for linear prediction in the forward direction using real points) using the same conventions we used earlier. Then, we multiply TD1 and TD2 by 1.5 to give us the extrapolated points we wish and we store those values as LPBin values for each axis. The remaining points that are not accounted for by LPbin or TD are zero filled automatically.

Now that we have all of the relevant values set, we are ready to process the spectrum using our new settings. This can be done using a built in command as well, simply XFB( ). However, let us assume that this command WASN’T hard coded into topspin. In this case, there is a lovely function called XCMD( ) where you simply type in the command you would use in the command line. In this case, we would use XCMD(‘xfb’) to perform this action.

After this, we have our new spectrum returned with linear prediction and zero filling performed. We could end there, but there is one more feature that you might like to know about. Using the built in functions, some of them can be passed variables or commands that alter the way the function is performed. Take, for instance, peak picking. If we were using this script to do automatic peak picking on the spectrum, the last thing we want is to have the peak picking dialog box pop up for each of our 100 samples - so we' disable the pop up box by instead opting for the silent mode of peak picking.

And voila!
As you can see, the python option allows you to manipulate data in a very similar manner to the Macro’s, but also allows for a bit more control. For instance, there are even options to browse through all your data to selectively process things. It also allows you to pull data into the script and compare directly - handy for dereplication if you have a library… I’ll post a tutorial in a few weeks showing exactly what I mean by that, as well as a lovely little function I’ve created that allows for semi-supervised peak picking of metabolomics data.
Although there does exist explicit documentation for using python in Topspin, I’ve found that I wish it had a list of all of the built in functions that was readily accessible. However, they do offer about 15-20 pages on selective use cases, so it’s a good start.