Macros are the most basic processing schemes available to a user in Topspin, and provide a great level of automation with very basic skills. Simply put, a well designed macro could automate a large portion of your workflow, allowing you to process data in your sleep.
It is overly simplistic, but for the sake of this quick start guide, think of each line of a macro as the command line at the bottom of Topspin. When you open your data, you use commands in this line for basic transformations of the data, editing processing variables, and even adjusting the viewing window. These commands can have variables passed to them in the command line as well, which is how we should think about them in a processing macro.
Lets take a simple example of 1D data – using only the command line to process it. Then, we’ll write a macro which will do the same transformations on the data, and finally, we’ll link it to the Topspin “serial” command to automate processing for multiple datasets.
(Find the dataset here)
Command Line Processing
When we open this dataset, we need to transform it first. For this, we use the line command “ft”. Once we have a spectrum, we can see that we need to apply phase correction. If we use automated phase correction, the command for 1D data is “apk”. Next, we perform baseline correction without integration by “absn”. Following these three commands, we have processed our data to the point an investigator might begin looking at the spectrum for peaks of interest. There are, of course, other commands and combinations – depending on what your processing scheme might be. As an example, if you wish to include integration into this scheme, you have three choices. You can either change the commands fed into the system – replacing “absn” with “abs” which uses automatic integration, you can implement integration in another step, or you can choose to integrate the spectra yourself. Hopefully, you can see the flexibility of having all three options available, depending on your application.
Since you have to perform these basic functions on every spectra, why not construct a macro that would do it for you with an easier command? These few seconds you save may not seem like much, but with even a 3 second savings per spectra, a sample size of 100 samples processed could save you more time than you spend writing the macro: so lets do that.
Writing and Editing Macros
First, you have to open up the Macro menu in Tospin by typing the command “edmac”. This launches the Macro menu, likely populated with a lot of Bruker provided scripts that automate a large chunk of processing. First, lets look one of the provided examples, the example_efp macro – open this up by highlighting the script and selecting edit.
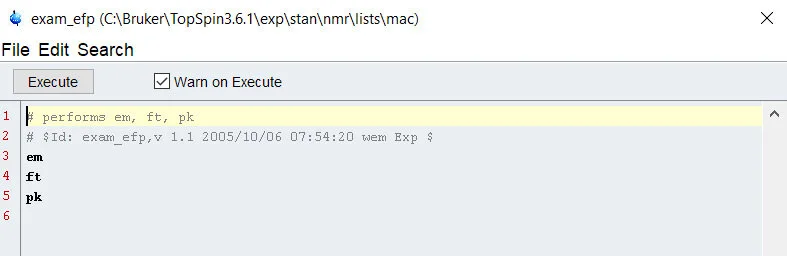
By selecting edit, you launch the macro edit utility, which is similar to a idle processor/notepad. By looking at this example, we can see that – much like python – we can write notes alongside commands by using the # sign before writing in a line. As the program moves down the line, these lines are ignored completely, allowing you to leave a detailed explanation of each step – or slip in some user information or metadata about the sample sets you are writing the macro for. Keeping highly detailed coding notes is a VERY SMART MOVE. The line structure of the Macro allows you to command the program to do one task, and when it is complete, it moves on to the next task. Dissecting the example script above, we can see that it uses a similar approach to basic processing:
· Perform exponential window multiplication with “em”
· Perform Fourier transformation with “ft”
· Perform phase correction with “pk”
For the sake of this quick start, we’re going to start fresh and write our own script. In order for us to edit or create a new macro, we need to change the source directory where topspin is looking for our macros. By default, this normally opens to the bruker directory (C:…..expt\stan\nmr\lists\mac) – to navigate to your directory, simply select the drop down menu and select the (…mac\user) version of the directory. If you’ve never experimented with Macros, this will be empty. Select File > New. Here, you’ll be prompted for a name, which can change later. For now, let’s name this something easy – ‘JE_tut1’.

Lets try writing a quick macro to do the commands we outlined on our 1D data – ft, apk, absn. Once we’re done, you can simply click execute to test the command – if it processes without flagging an error, it worked!
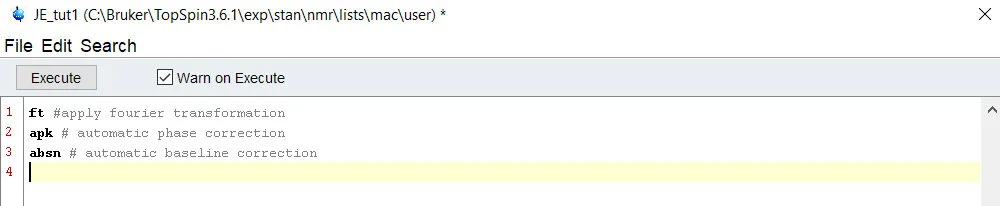
If you’re satisfied with the macro, you can save it and recall it any time with a variety of different methods. My favorite, is the ability to call on a macro/script/python script by simply using it’s name in the command line. Try it by saving the script, exiting out of the macro window, and typing “JE_tut1” in the command line. Alternatively, you can launch the macro by using the command “xmac[space]name_of_macro” – this is helpful if you have different versions of a script floating around – such a Macro and Python script both called ‘process 1D’.
Partnering Macros with serial
Macros, scripts, and python scripts are great time savers, but the real power comes when you can automate processing on more than one spectra at a time. Topspin has a built in function to do this called serial that allows you to perform a single task on many spectra at a time.
Step 1: Define the list of samples to process
In topspin 3, select process spectrum > advanced > serial (or simply type ‘serial’ into the command line). From there, you’ll see all three options: define list, define command, execute. For ease of use, we’ll be using the option to ‘build dataset list using find’.
Step 2: Using “find” to build a dataset list
Launching this window, you’ll see lots of different methods of filtering data; name, experiment number, pulse program, etc.. we’ll start by applying our 1D NMR quick script to a large sample subset of 1D data. To do this, we filter all of the data in our data directory using the pulse program ‘zg’. This returns a list of all the experiments in the selected directory(ies) that fit that pulse program – however, you may notice it does a simple string search and you will get results from any pulse program that contains the characters you searched for. Be sure you only select the datasets that use the ‘zg’ pulse, not – for instance – a ‘zgpg’. Once you hit ‘ok’ you’ll see a message at the bottom telling you where it saved the list – if you’d like, you can recall this list later – but you should copy it from the TEMP folder and rename it something easier to remember.
Step 3: Define Command
The last thing to do is define the command you wish to execute on all of the selected data sets. Since we wrote a macro to process all of the 1H spectra using zg, we will apply this macro here by typing JE_Tut1 as the command – remember, you can call scripts/macros directly by name!
Execute the macro – and watch it work! If you’re sitting on 100 spectra, it will chug through these in order until it’s complete. Perfect to set up right before that meeting you have down the hall.
Expanding Macros to suit your needs
You can add other features into the macros as well, such as the ability to zoom into certain regions of a spectra, peak picking in only one region, and more. Lets look at a more complex example here – NUS 2D HSQC data. There are a few more things we need to consider when looking at 2D data, as well as NUS data processing. For the purposes of this tutorial, I’m not going to get into things like linear prediction or zero filling – but these are completely automatable using macros. Instead, there are a few complications with these data having been NUS collected, so we will keep those in and you can read up on linear prediction on your own.
This script also uses arguments, which are simply provided by following the command with a space and then the argument value you are setting. As an example, if we were changing the “SI” of a processed spectrum, we can set it by:
“Command value1 value2”
“SI 4k 1024”
When working with NUS data, there are ‘holes’ in the data – it needs a special kind of reconstruction. Since Topspin 3.5pl6, there are reconstruction algorithms that are provided for use without a special license. However, if you’ve been processing NUS data, and have seen a little error message that pops up telling you only have access to the free reconstruction techniques, we can get rid of that in our macro.
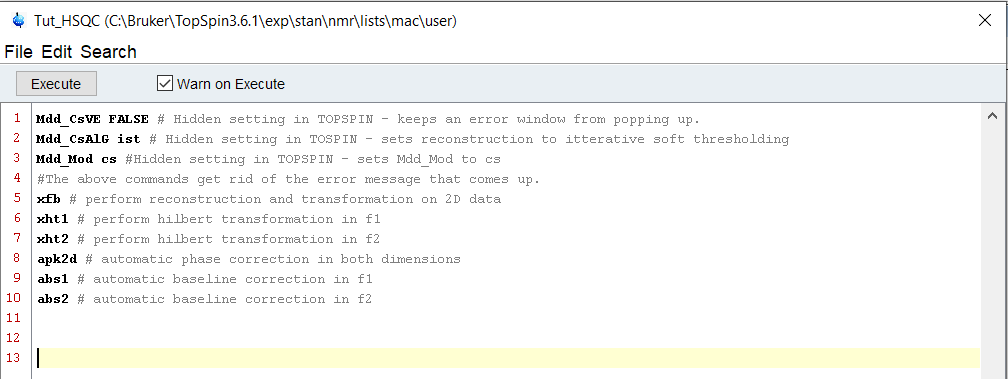
We’ve woven in a couple of small QOL features in this macro that save us a few clicks and a few seconds per spectra. For instance, we are not able to phase correct the spectra if we do not have the imaginary part of the spectrum, which we do not collect in NUS data – so we calculate it with the Hilbert transformations for each axis. Once that is done, it’s simple to do phase correction and have a good starting point for analysis.
So there you have a quick entry into the world of Topspin macros and two small examples to get you going. Remember, there’s extensive documentation on how to automate your processing with topspin in the manuals section. By combining these simple macros with the serial command, you can quickly optimize your NMR processing for many datasets at a time.