Automation scripts in Topspin are coded as C language based scripts. This differs from python style scripts significantly, and has its advantages and drawbacks like any programming language. Without making too many comments on preferences, I will say that I do not know much about C languages, except some basic differences (such as having to define what kind of value a variable will stand for, explicitly). As is the case with python and macro scripts, you will need to navigate to the edit screen to find these codes - but this time with edau.
Commands are typed in bold - edpy/edmac/edau
Variables are in italics - i1
The good news is that much like the macros and python, many commands that you would use in topspin are already coded in as commands you can call directly. This means that if you wanted to clone our macro into an automation script, you can do that! In fact, a few of the commands you may already use already ARE automation scripts! As an example, lets pull up one of the many provided scripts, multizg.
If you are using a spectrometer without IconNMR or the spooler service activated, you may be familiar with multizg.. but what does it actually do? Navigate to the Burker script directory with edau and find the multizg script.
From the description in the au file, you can see that multizg program written by Rainer Kerssebaum contains a few blocks of code to allow for multiple experiment acquisition. If you have set up experiments beforehand (10 -1H , 11-HSQC, 12-TOSY) then when you activate multizg and enter in the correct number of experiments, it will read each file and run the acquisition. If you haven’t set up the experiments beforehand though, it will simply copy the open acquisition and run that many of that particular experiment – this has caught me a couple of times...
If you look at the way it iterates through the experiments, it prompts the user for the value of ‘i1’ which can be thought of as the number of experiments to be run. If i1 > 0, as it should be, then it calculates the number of experiments that are queued up and the experiment time for each. Once that has been completed, it iteratively runs through the experiments until all of them have been completed. Pretty cool right? Now, this is a very advanced script, and it looks pretty dense to a new user – DON’T PANIC. Lets try to write a very simple Au script to do some 1D processing. As you get more comfortable, try writing some more cleaver Au scripts to perform your tasks for you!
Sticking to the command line arguments, simply write a new Au script called 1D_ZG. Assuming we’ve set up the experiment correctly and it is open, let’s write a script to set the receiver gain and then acquire the data.
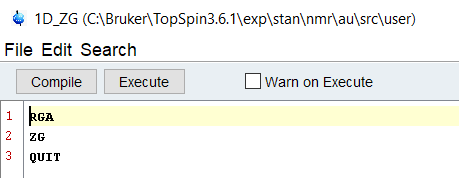
Unlike Macros and Python scripts, Au scripts must be compiled before they can be executed. Select “Compile” and after a few seconds, you will get a prompt that tells you it’s ready to go! If you have errors or language in there that confuses the system, it will give you a kickback message that tells you something is incorrect. Au scripts offer the users who are familiar with C based languages a chance to put their skills to work for them, and it’s a fantastic option. Try looking through some other provided scripts, such as the pulsecal script, to get a better idea of how to interact with Topspin.
If you were lucky enough to catch the advanced Topshim webinar by Bruker, you might have seen how you can further expand our script to include specific shimming steps as well, further automating your acquisition step. As is the case with all three types of automation, once you have these scripts established in your directory, you can simply call them up using the command line - or even code in your own buttons!